How does polymerase chain reaction work step by step?
PCR works by copying a selected piece of DNA through repeated heating and cooling. A laboratory first extracts genetic material from a sample, mixes it with primers, nucleotides, DNA polymerase, and buffer, then places the mixture inside a thermal cycler. The machine separates the DNA strands, lets primers attach to the target, and gives the polymerase time to build new strands. Repeating this cycle can turn a tiny amount of DNA into millions or billions of detectable copies.
The basic idea sounds simple, yet each stage has a specific job. A poor sample, badly designed primers, contamination, or the wrong temperature can change the result. This guide explains how PCR works step by step, what happens inside the reaction tube, and how the amplified DNA is detected after cycling.
What is PCR?
Polymerase chain reaction, commonly called PCR, is a laboratory method used to make many copies of a chosen DNA sequence. It allows scientists to study genetic material that may be present in quantities too small for direct examination.
A biological sample can contain a huge mixture of DNA. Only a tiny part may be relevant to the test. PCR does not copy every sequence at random. It uses two short pieces of synthetic DNA, called primers, to mark the beginning and end of the region that should be copied.
Once the target has been selected, repeated temperature changes drive the copying process. Each successful cycle creates new templates that can be copied during the next cycle.
This repeated copying is why PCR is called a chain reaction.
What does PCR need to work?
A standard PCR reaction needs template DNA, two primers, a heat-stable DNA polymerase, nucleotides, magnesium ions, buffer, and purified water. Each part supports a different stage of DNA copying.
Template DNA
Template DNA is the genetic material that may contain the sequence being tested. It can come from blood, saliva, tissue, cultured cells, plants, bacteria, food, soil, or many other sample types.
PCR can sometimes begin with only a small amount of template. Still, the DNA must be clean enough for the enzyme to work.
Chemicals left behind during extraction may block the reaction. Blood components, alcohol, salts, detergents, plant compounds, and substances found in soil are common PCR inhibitors.
Forward and reverse primers
Primers are short, single-stranded DNA sequences designed to bind on opposite sides of the target region. One is called the forward primer, while the other is called the reverse primer.
Together, they define the DNA fragment that will be copied.
Primer design affects specificity. A primer that also matches unrelated DNA may produce unwanted fragments. A primer pair that does not bind well may produce a weak signal or no product at all.
Heat-stable DNA polymerase
DNA polymerase is the enzyme that builds new DNA strands. Standard PCR often uses Taq polymerase, an enzyme originally isolated from the heat-loving bacterium *Thermus aquaticus*.
Ordinary enzymes may lose their shape when exposed to high temperatures. Taq polymerase can survive the repeated heating steps used during PCR.
Some reactions use other polymerases. High-fidelity enzymes are selected when copying accuracy matters, such as during cloning or sequencing preparation. Hot-start enzymes remain largely inactive during reaction setup and become active after heating, which can reduce unwanted amplification.
Deoxynucleotide triphosphates
Deoxynucleotide triphosphates, usually shortened to dNTPs, are the building blocks used to create new DNA.
The four types are:
- dATP, which contains adenine
- dTTP, which contains thymine
- dCTP, which contains cytosine
- dGTP, which contains guanine
DNA polymerase adds these nucleotides in an order guided by the template strand.
Magnesium, buffer, and water
Magnesium ions are needed for DNA polymerase activity. Too little magnesium may produce weak amplification. Too much may increase unwanted products.
The buffer keeps the reaction at a suitable pH and provides the chemical conditions required by the enzyme. Nuclease-free water brings the reaction to its planned volume without adding enzymes that might damage DNA.
How does PCR work step by step?
PCR begins before the tube enters the thermal cycler. Sample collection, nucleic acid extraction, reagent preparation, cycling, and product detection all contribute to the final result.
Step 1: The biological sample is collected
The first step is collecting a sample that may contain the target genetic material. The collection method depends on what the laboratory wants to detect.
A respiratory test may begin with a nasal or throat swab. A genetic test may use blood, saliva, or cheek cells. Food testing may begin with a small portion of the product, while environmental testing may use water or soil.
Collection quality can affect everything that follows. A swab that does not gather enough material may lead to a false-negative result. Poor storage can also damage nucleic acids before testing begins.
Samples are often stored at controlled temperatures and transported in containers designed for the specimen type. The exact requirements depend on the assay and the stability of the target.
Step 2: Cells or particles are broken open
DNA is usually enclosed within cells, nuclei, bacteria, or other biological structures. The sample must be treated so that the genetic material becomes accessible.
This stage is commonly called lysis.
Lysis may involve detergents, enzymes, heat, mechanical force, or a combination of methods. Detergents can break cell membranes. Protein-digesting enzymes can help remove proteins attached to DNA.
Viral samples need a method suited to the virus being tested. Some viruses have a lipid envelope, while others have a tougher outer structure.
Step 3: DNA or RNA is extracted and purified
After lysis, the genetic material is separated from proteins, lipids, salts, and other sample components. The goal is to produce a clean nucleic acid solution that can be added to the PCR mixture.
Many laboratories use silica columns or magnetic beads. Under selected chemical conditions, DNA or RNA binds to the solid surface. Unwanted material is washed away, and the purified nucleic acid is released into a clean liquid.
Extraction is more than a preparation step. Poor recovery can leave too little target for detection. Incomplete washing may leave inhibitors in the sample.
Standard PCR directly copies DNA. When the starting material is RNA, it must first be converted into complementary DNA, or cDNA, by an enzyme called reverse transcriptase. This version of the method is called reverse transcription PCR.
Step 4: The PCR master mix is prepared
The extracted template is combined with the primers, DNA polymerase, dNTPs, magnesium, buffer, and water. Laboratories often prepare a master mix containing all shared reagents before dividing it among reaction tubes or wells.
A master mix helps reduce pipetting differences between samples. It also lowers the number of separate liquid transfers.
The template is usually added after the shared mixture has been distributed. Positive controls, negative controls, and patient or research samples are kept clearly separated.
Clean technique matters during this stage. PCR can copy very small amounts of DNA, including DNA introduced by accident. A droplet from an earlier reaction may contain enough amplified product to contaminate a new test.
Laboratories commonly separate reagent preparation, sample handling, and post-amplification work into different areas. Filtered pipette tips, clean benches, gloves, and regular surface cleaning also help control contamination.
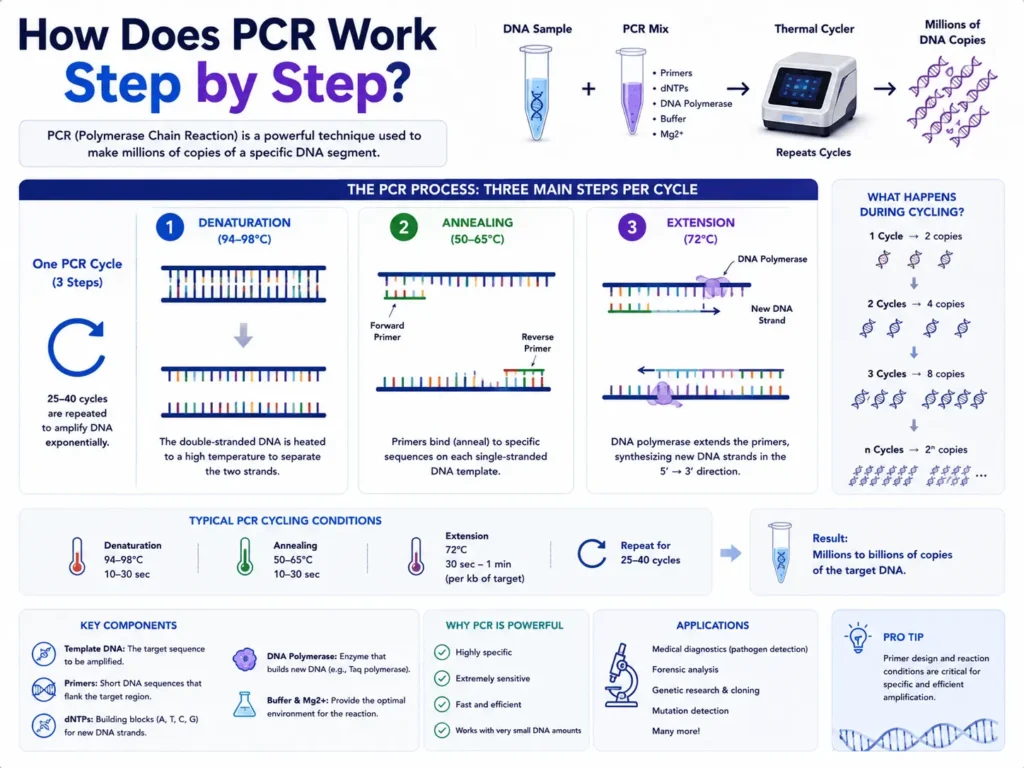
Step 5: Initial denaturation opens the DNA and activates the reaction
The prepared tubes are placed in a thermal cycler. This instrument changes the temperature according to a programmed schedule.
Many protocols begin with an initial denaturation step near 94°C to 98°C. The reaction may remain at this temperature for several seconds or a few minutes.
Heat breaks the hydrogen bonds holding the two DNA strands together. The double-stranded DNA becomes two single strands that can serve as templates.
Some hot-start polymerases also need this first heating period for activation. The required temperature and duration depend on the enzyme.
Initial denaturation should be long enough to separate the template. Excessive heating may damage DNA or reduce enzyme performance, especially during long programs.
Step 6: Denaturation separates the DNA strands during each cycle
After the first heating stage, PCR enters its repeated cycling program. Denaturation is usually the first stage of every cycle.
The reaction is commonly heated to around 92°C to 98°C for a brief period. At this temperature, newly formed double-stranded DNA separates again.
Each strand now carries the sequence that the primers can recognize. Both original DNA and products created in earlier cycles can serve as templates.
DNA with a high proportion of guanine and cytosine bases may need stronger denaturation conditions because GC base pairs form three hydrogen bonds. Adenine-thymine pairs form two.
Poor denaturation can reduce the amount of target copied. Excessive temperature or prolonged heating can weaken polymerase activity over many cycles.
Step 7: Annealing allows the primers to bind
The thermal cycler lowers the temperature so the primers can attach to complementary sequences on the single DNA strands. This stage is called annealing.
Annealing temperatures often fall between 50°C and 65°C, though some assays run outside this range. The correct value depends mainly on primer sequence, primer length, salt conditions, and melting temperature.
The forward primer binds to one template strand. The reverse primer binds to the opposite strand on the other side of the target.
Annealing temperature affects which sequences get copied. A temperature that is too low may allow primers to bind imperfectly to unrelated regions. This can create extra PCR products.
A temperature that is too high may stop the primers from binding efficiently. The result may be a faint product or complete reaction failure.
Primer-dimers can also form during this stage. These small unwanted products appear when primers bind to each other and are copied by the polymerase.
Step 8: Extension builds new DNA strands
The temperature rises to a level suited to the DNA polymerase. Taq polymerase commonly works near 72°C, although the preferred temperature varies between enzymes.
DNA polymerase begins at the attached primer and adds nucleotides to its 3′ end. It reads the template strand and builds a complementary strand in the 5′ to 3′ direction.
Adenine pairs with thymine. Cytosine pairs with guanine.
The time allowed for extension depends on the enzyme and the length of the target. Short fragments may need only a few seconds. Longer products need more time.
At the end of this stage, each starting double-stranded DNA molecule has been copied into two double-stranded molecules, assuming the reaction performs with perfect efficiency.
Step 9: Denaturation, annealing, and extension are repeated
The thermal cycler repeats the three main stages:
- Denaturation separates the strands.
- Annealing lets the primers bind.
- Extension creates new strands.
A standard program may run for about 25 to 40 cycles. Thirty to 40 cycles are common in many diagnostic and research assays, though the proper number depends on the method.
During the earliest cycles, the target may still be too scarce to detect. Each new product then becomes a template in later cycles.
Under ideal conditions, the number of target copies doubles with each cycle. Starting from one copy, 30 perfect cycles would produce about 1.07 billion copies because the theoretical copy number follows 2³⁰.
Real reactions rarely maintain perfect doubling through every cycle. Reagent limits, enzyme activity, product competition, and inhibitors reduce efficiency as amplification continues.
Step 10: Final extension completes unfinished products
Many conventional PCR programs include a final extension near the end of cycling. The reaction may be held near the polymerase’s preferred extension temperature, often around 72°C, for several minutes.
This gives the enzyme time to finish strands that were not fully extended during the final cycle.
A final extension can be useful when complete, full-length products are needed for gel analysis or later laboratory work. Its value depends on the polymerase and assay design.
Some modern protocols use very short targets or combined annealing-extension stages and may not need a separate final extension.
Step 11: The reaction is cooled
After cycling, the thermal cycler usually lowers the temperature to about 4°C to 12°C. This temporary hold helps preserve the amplified DNA until the tubes are removed.
Cooling does not create more target copies. The main amplification process has already ended.
PCR products should still be handled carefully after the run. Opening tubes can release concentrated amplified DNA into the work area. This material can contaminate later reactions.
Closed-tube systems, including many real-time PCR assays, reduce the need to open amplified samples.
Step 12: The amplified DNA is detected
PCR amplification alone does not always provide a visible result. The laboratory needs a way to determine whether the target product was created.
The detection method depends on the type of PCR.
Detection after conventional PCR
Conventional PCR products are commonly examined with agarose gel electrophoresis, which separates nucleic-acid fragments by size. The amplified DNA is loaded into a gel, and an electric field moves the negatively charged DNA fragments through it.
Smaller fragments generally move farther than larger fragments. A DNA ladder provides size markers for comparison.
A fluorescent DNA-binding dye makes the bands visible under suitable light. A band at the expected position suggests that a product of the planned size was created.
A band does not automatically prove that the sequence is correct. Unwanted products can sometimes have a similar size. Restriction analysis or DNA sequencing may be used when stronger confirmation is needed.
Detection during real-time PCR
Real-time PCR, also called quantitative PCR or qPCR, measures fluorescence while amplification is taking place.
The signal may come from a dye that binds to double-stranded DNA or from a sequence-specific fluorescent probe. As the target accumulates, fluorescence rises.
The instrument records the cycle at which the signal crosses a set threshold. This value is often called the quantification cycle, or Cq. Some systems use the term cycle threshold, or Ct.
A sample with more starting target usually crosses the threshold earlier than a sample with less starting target. Cq values should be read through the rules of the assay rather than treated as universal measurements.
Why does PCR amplification become exponential?
PCR can create a large amount of DNA because products from one cycle become templates in the next. The target population can grow rapidly while reagents remain available and the reaction remains efficient.
One target molecule can become two after one ideal cycle. Those two can become four, then eight, 16, 32, and so on.
The theoretical relationship is:
Number of copies = starting copies × 2ⁿ
In this expression, n is the number of cycles.
This equation describes perfect amplification. Actual reactions usually produce fewer copies because efficiency is below 100%.
Amplification also reaches a plateau. Primers and nucleotides become limited, polymerase activity falls, and accumulated products begin to compete with primers for binding.
How do primers make PCR target-specific?
Primers control which part of the DNA is copied. Their sequences are chosen to match regions flanking the target.
DNA polymerase cannot begin a new strand from nothing. It needs a primer with a free 3′ end.
When both primers bind in the correct places and face toward each other, the sequence between them becomes the amplicon. The amplicon is the DNA fragment produced by PCR.
Specificity depends on more than primer sequence. Annealing temperature, magnesium concentration, primer concentration, template quality, and cycle number can all affect unwanted binding.
A well-designed assay may also use a probe that recognizes a sequence inside the amplicon. This adds another layer of target recognition in many real-time PCR methods.
How is RNA tested when PCR copies DNA?
PCR polymerases copy DNA rather than RNA. An RNA target must be converted into cDNA before standard amplification can begin.
Reverse transcriptase builds a DNA strand using RNA as its template. The resulting cDNA enters the usual PCR cycle of denaturation, annealing, and extension.
This method is called reverse transcription PCR, commonly written as RT-PCR.
RT-PCR is used in gene-expression studies and in tests for RNA viruses. It should not be confused with real-time PCR. A test can be reverse transcription PCR, real-time PCR, or both.
When RNA conversion and real-time detection are combined, the method may be described as real-time RT-PCR or RT-qPCR.
What controls are used in PCR?
Controls help a laboratory tell the difference between a true result and a failed or contaminated reaction. A PCR run without suitable controls may be hard to trust.
Positive control
A positive control contains the target sequence or another approved target material. It should produce amplification.
Failure of the positive control may point to a reagent problem, incorrect cycling conditions, enzyme failure, or an instrument issue.
No-template control
A no-template control contains the reaction mixture but no sample DNA. Water usually replaces the template.
It should remain negative. Amplification in this tube may point to contaminated reagents, unwanted primer products, or DNA introduced during setup.
Negative extraction control
A negative extraction control passes through the extraction process without containing the target. It can reveal contamination introduced during extraction.
Internal amplification control
An internal control is amplified in the same tube or alongside the sample. It helps show whether extraction worked and whether inhibitors blocked the reaction.
A sample that produces no target signal and no internal-control signal may be invalid rather than truly negative.
What can cause PCR to fail?
PCR may fail because of poor sample collection, damaged DNA, inhibitors, incorrect reagent amounts, unsuitable temperatures, primer problems, or contamination.
No amplification may appear when the template is absent or too scarce. It can also happen when the annealing temperature is too high, the polymerase is inactive, or a required reagent was not added.
Multiple bands often point to nonspecific primer binding. A low annealing temperature, excessive magnesium, high primer concentration, or too many cycles may contribute.
Smearing on a gel can result from degraded DNA, excessive template, unwanted amplification, or overloaded gel wells.
False-positive findings may come from amplified DNA carried over from an earlier run. False-negative findings may occur when the target is present but extraction is poor or inhibitors block amplification.
Careful controls, validated procedures, and separate clean work areas help laboratories recognize these problems.
How long does PCR take?
The amplification program commonly takes about one to three hours. The full testing process may take longer once sample preparation, extraction, result analysis, and reporting are included.
Run time depends on the number of cycles, tube format, thermal cycler speed, target length, and enzyme.
Fast-cycling instruments can change temperatures quickly and may use short holding times. Some assays combine annealing and extension into one stage, creating a two-step cycling program.
A rapid machine does not remove the need for careful sample handling. The result is only as dependable as the full process behind it.
What is PCR used for?
PCR is used whenever a small, selected DNA or RNA target needs to be detected, copied, measured, or studied.
Medical laboratories use PCR to test for infectious organisms and selected genetic changes. Research laboratories use it to study genes, prepare DNA for sequencing, check cloned fragments, and examine gene activity through reverse transcription methods.
PCR also supports forensic analysis, food testing, animal health, plant disease testing, ancestry research, and environmental monitoring.
The core copying process remains similar across these uses. The sample, primers, detection chemistry, controls, and rules for reading the result change with the assay.
Does a positive PCR result always mean active disease?
A positive PCR result means the assay detected its genetic target under the test’s stated rules. The medical meaning depends on the specimen, timing, target, assay performance, and clinical setting.
PCR may detect genetic material even when an organism is no longer capable of causing an active infection. In other cases, a low target level may reflect an early infection, a resolving infection, contamination, or material near the assay’s detection limit.
A negative result also has limits. The target may be absent, below the detection limit, collected from the wrong site, or lost during storage and extraction.
Clinical PCR findings should be read alongside symptoms, exposure history, specimen quality, controls, and any other relevant tests.
PCR turns an invisible DNA target into measurable evidence
PCR works through a controlled sequence: collect the sample, release and purify the nucleic acid, prepare the reaction, separate the DNA strands, bind target-specific primers, extend new strands, and repeat the cycle.
Its strength comes from repetition. Every successful cycle supplies more templates for the next one, allowing a tiny starting amount to become detectable.
The machine performs the temperature changes, but dependable PCR still rests on good primer design, clean sample handling, suitable controls, and careful reading of the data. Once those pieces are in place, the reaction provides a clear way to study genetic material that would otherwise remain too scarce to see.