Why Do Some qPCR Amplification Curves Look Abnormal?
Some qPCR amplification curves look abnormal because the reaction is no longer behaving like a clean, target-specific PCR. The cause may be simple, such as a bad baseline setting, pipetting variation, low template amount, or a weak fluorescent signal. It may also point to a deeper problem, such as PCR inhibitors, primer-dimers, nonspecific products, degraded sample material, contamination, or poor assay design.
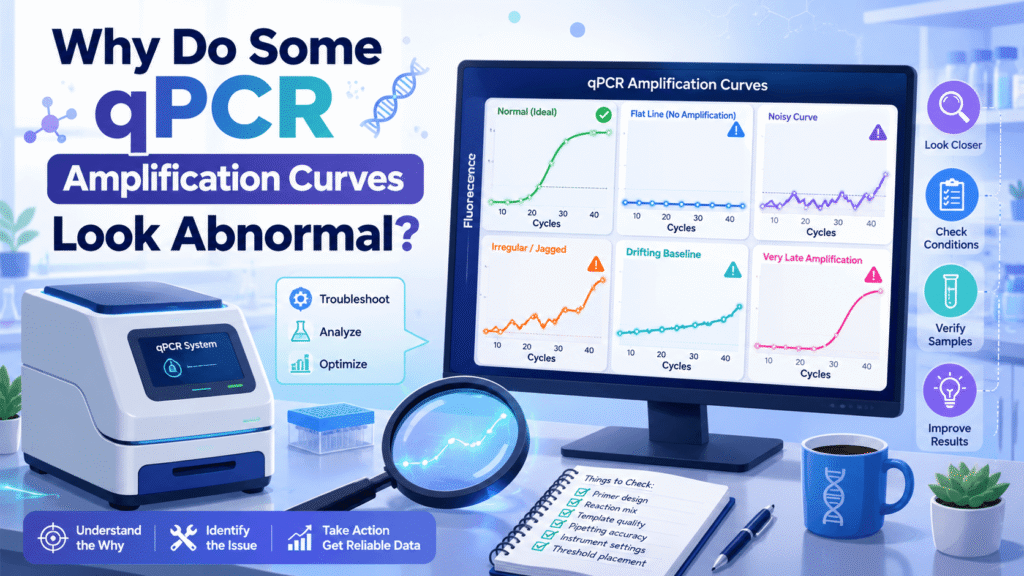
A normal qPCR curve should usually show a flat baseline, a clear exponential rise, and a plateau. When that shape changes, the curve is giving you a warning. Sometimes the result can still be saved by checking the log view, melt curve, NTC wells, and replicate spread. Other times, the run should be repeated because the Cq value cannot be trusted.
Abnormal curves are not random noise most of the time. They often follow patterns. Once you learn what each pattern means, troubleshooting becomes much less stressful.
What does a normal qPCR amplification curve look like?
A normal qPCR amplification curve has three main phases: baseline, exponential amplification, and plateau. The baseline should stay flat at the start, the exponential phase should rise cleanly, and the plateau should appear after reagents begin to run out.
During the early cycles, the fluorescent signal is usually too low to separate from background noise. A clean baseline gives the software a fair starting point for judging when real amplification begins.
Once enough product builds up, fluorescence rises above background. The threshold is usually placed within the exponential phase, because this is the part of the curve where amplification is most consistent. In qPCR workflows, the amplification threshold is usually based on background fluorescence from the baseline region, then used to mark where reporter signal rises above that background level.
The shape matters because qPCR is not only about whether a target appears. It is also about when the signal crosses the threshold.
A clean curve usually has:
- A stable baseline in early cycles
- A smooth exponential rise
- Tight replicate curves
- A clear plateau
- No amplification in no-template controls
- A melt peak or probe signal that matches the expected product
When one of these parts looks wrong, the curve may still cross the threshold, but the number can be misleading.
Why do qPCR curves rise too late?
qPCR curves rise too late when the starting target amount is low, the sample is partly degraded, the reaction is inefficient, or something in the sample is slowing PCR. A late Cq is not always wrong, but it needs careful checking.
A true low-copy target can naturally amplify late. This often happens with weakly expressed genes, low pathogen load, diluted samples, or old extracts with little usable nucleic acid left.
The problem begins when late curves look weak, jagged, or inconsistent across replicates.
PCR inhibitors are a common reason. Substances carried over from extraction can slow polymerase activity. These may include ethanol, salts, heme, humic acids, phenol, detergents, or high sample input. In multiplex qPCR, one target can also suffer when another target is more abundant or more efficient.
In multiplex reactions, an abundant or highly efficient target can use up shared reaction components, which may delay a weaker target and make its curve appear late.
Late curves also appear when primers are not binding well. This can happen if the annealing temperature is too high, primer design is poor, or the target sequence has mismatches near the primer or probe binding region.
A late curve should be reviewed with the controls. If the positive control amplifies normally but the sample is late, the issue may sit inside the sample. If all wells amplify late, the master mix, cycling program, primer concentration, or instrument settings may be the cause.
Why do qPCR curves show no amplification?
No amplification means the fluorescence never rises above the threshold in a meaningful way. This can happen because the target is absent, the template is degraded, a reaction component is missing, or the wrong detection channel was selected.
No curve is not always a failure. In a true negative sample, no amplification is the expected result.
The concern begins when a positive control fails, a known positive sample stays flat, or some replicates amplify while others do not.
A flat qPCR curve may happen because of:
- No target sequence in the sample
- Too little template added
- Degraded DNA or RNA
- Failed reverse transcription in RT-qPCR
- Missing polymerase, primers, probe, MgCl₂, or master mix
- Incorrect cycling program
- Wrong dye or detector setting
- Poor plate sealing or evaporation
- Instrument reading issue
No amplification often traces back to practical setup problems, such as missing template, absent target, missing reaction components, degraded sample, or incorrect dye detector settings.
When no amplification appears, the first check should be the positive control. If the positive control also fails, the run has a setup or reagent problem. If the positive control works, the sample itself may be negative, inhibited, degraded, or below the detection limit.
Why do qPCR curves amplify in no-template controls?
Amplification in a no-template control usually means contamination or primer-dimer formation. This is one of the clearest warning signs in qPCR because the NTC should not contain target nucleic acid.
A no-template control contains all reaction components except sample template. If it amplifies, the signal came from somewhere else.
The most serious cause is target contamination. This can come from previous PCR products, positive-control material, shared pipettes, aerosols, contaminated water, or a reagent that was exposed during setup. Exponential amplification in NTC wells often points toward carryover contamination, especially when the same target sequence has been handled in the lab before.
The other common cause is primer-dimer formation. In SYBR Green assays, any double-stranded DNA can produce fluorescence. That means primer-dimers may create a curve even when no true target exists.
Melt curve analysis becomes especially useful in SYBR Green qPCR because the dye reacts with any double-stranded DNA, not only the intended target. Melt curves help confirm whether the expected qPCR product formed because each double-stranded product separates at a characteristic temperature during heating.
If the NTC amplifies very late and has a low-temperature melt peak, primer-dimer is likely. If it amplifies near the sample Cq and has the same melt peak as the target, contamination is more likely.
That difference changes the next step. Primer-dimers call for assay redesign or reaction adjustment. Contamination calls for cleaning, reagent replacement, workflow separation, and stricter handling.
Why do qPCR curves look jagged or noisy?
Jagged qPCR curves usually come from weak fluorescence, bubbles, poor optical contact, dirty plates, evaporation, baseline errors, or very low target concentration. Noise becomes more visible when the true signal is near the assay’s limit.
A jagged curve is not the same as a clean exponential curve. It may cross the threshold, but the crossing point may not reflect real amplification.
Small bubbles are a surprisingly common cause. A bubble in the well can scatter light and create sudden signal jumps. Fingerprints, dust, scratches, poor sealing film, or condensation can also interfere with fluorescence reading.
Low reaction volume can make the signal less stable. Evaporation can make this worse, especially around plate edges or in poorly sealed wells.
Jagged curves also happen near the limit of detection. At very low copy number, stochastic effects become more visible. One replicate may receive a target molecule while another receives none. The result is a messy cluster of late curves.
The best first step is to look at the raw amplification plot and the log view. For cleaner analysis, amplification curves are commonly reviewed in log plot view, with the threshold placed near the middle of the exponential phase.
If the curve only looks abnormal in linear view but appears clean in log view, the issue may be display scaling rather than biology.
Why do qPCR replicates look different from each other?
qPCR replicates look different when reaction setup, sample quality, template amount, or amplification efficiency varies between wells. Tight replicates suggest the assay is stable. Wide replicate spread means something in the workflow needs attention.
Replicate variation often comes from pipetting. qPCR reactions use small volumes, so even a tiny difference can affect the final Cq.
Poor mixing is another cause. If template or master mix is not evenly mixed, one well may receive more target or primer than another.
Replicates can also separate when the target is near the detection limit. At low copy number, random distribution of molecules becomes a real issue. One well may amplify, another may amplify late, and another may stay flat.
Other causes include:
- Bubbles in some wells
- Edge evaporation
- Uneven sealing
- Inhibitor carryover in one sample aliquot
- Poorly mixed cDNA
- Degraded RNA
- Plate loading errors
- Different threshold behavior across wells
Replicate spread should not be fixed by simply deleting the ugly curve. A curve should only be removed when there is a clear technical reason, such as a visible bubble, failed seal, or known pipetting mistake.
Why do qPCR curves have a strange baseline?
A strange baseline appears when early-cycle fluorescence is unstable, the baseline range is set incorrectly, or background signal differs between wells. Since the threshold depends on baseline behavior, baseline problems can distort Cq values.
Baseline drift can make a curve appear to rise before real amplification starts. It can also hide early amplification if the baseline is set too wide.
Automatic baseline settings are helpful, but they are not perfect. In some assays, especially high-template samples, amplification may begin earlier than expected. If the software includes early amplification cycles inside the baseline calculation, the final Cq may become inaccurate.
A baseline that slopes upward may come from background fluorescence, dye instability, optical noise, or plate reading issues. A baseline that jumps suddenly may point to bubbles, condensation, or debris.
The threshold should sit in the exponential phase, not in the baseline noise and not in the plateau. The threshold should be checked in log plot mode so it sits within the true exponential phase rather than baseline noise or plateau signal.
When the baseline is wrong, the curve may look abnormal even if the chemistry worked.
Why do qPCR curves plateau too early or too low?
qPCR curves plateau early or too low when reaction components become limited, amplification efficiency drops, or the fluorescent signal is weak. The plateau phase is less useful for quantification than the exponential phase, but it can still reveal reaction trouble.
A low plateau may happen when there is not enough probe, primer, dNTPs, MgCl₂, or polymerase activity. It may also appear when inhibitors reduce amplification.
In some cases, a low plateau is not a major issue if the exponential phase is clean and the Cq is reliable. qPCR quantification depends mainly on the threshold crossing during exponential amplification.
Still, a strange plateau should not be ignored. It may hint at poor assay efficiency or inconsistent reagent performance.
PCR does not continue forever. Eventually, amplification slows. The PCR plateau can occur when primers become depleted, so a curve may stop rising even before every other reagent is fully exhausted.
A plateau that appears much earlier than expected may mean the reaction started with very high template input, reached reagent limits quickly, or produced nonspecific products that consumed primers.
Why do qPCR curves cross the threshold too early?
Early threshold crossing usually means high starting template amount, contamination, threshold misplacement, or unexpected amplification. If a sample amplifies much earlier than expected, it should be checked before being treated as a strong positive.
Sometimes an early Cq is real. A highly expressed gene, abundant pathogen target, or concentrated DNA sample can amplify early.
But an unusually early curve can also come from contamination. This is especially true if negative controls amplify, if the same target was handled recently in the lab, or if positive-control material was opened near sample setup.
Threshold placement can also create false early Cq values. If the threshold is placed too low, it may cut through baseline noise rather than true exponential amplification.
An early curve should be checked against the curve shape. A real early amplification curve should still have a smooth exponential rise. If the curve jumps, slopes oddly, or crosses the threshold during noisy baseline cycles, the Cq may be unreliable.
Why do qPCR curves show primer-dimers?
Primer-dimer curves appear when primers bind to each other and extend, creating small double-stranded products. This is especially visible in SYBR Green qPCR because the dye binds to any double-stranded DNA.
Primer-dimers often amplify late because they start from weak primer interactions rather than true template.
They are more likely when primer concentration is too high, annealing temperature is too low, primers have complementarity at the 3′ ends, or the assay runs for too many cycles.
Nonspecific products and primer-dimers are often tied to excessive primer concentration, too much magnesium, impure water, or contaminants in dNTPs. A common primer concentration range for PCR reactions is around 0.2–1 µM.
Primer-dimers can confuse results in two ways.
First, they may create false amplification in NTC wells. Second, they may compete with the target reaction and reduce sensitivity.
Melt curve analysis is the easiest way to catch primer-dimers in SYBR Green assays. A primer-dimer peak usually appears at a lower melting temperature than the true amplicon. Probe-based assays are less affected because signal depends on probe cleavage or binding, not just any double-stranded product.
Why do qPCR curves show nonspecific amplification?
Nonspecific amplification happens when primers bind to unintended sequences and create products that are not the target. The curve may still look real, but the signal does not represent the intended sequence.
This is one of the reasons a clean-looking qPCR curve can still be wrong.
Nonspecific amplification may appear when primer design is weak, annealing temperature is too low, magnesium concentration is too high, or the sample contains related sequences.
In SYBR Green assays, nonspecific products are especially risky because the dye cannot tell the correct amplicon from the wrong one. Melt curve analysis helps reveal this. Multiple melt peaks usually mean more than one product formed.
Gel electrophoresis can also help when melt curve results are unclear. A single band at the expected size supports specificity. Extra bands suggest nonspecific amplification.
Hot-start PCR can reduce nonspecific products because it limits primer extension from low-temperature interactions before the first denaturation step.
The best fix is usually assay redesign rather than repeated tweaking. Better primers often save more time than repeated troubleshooting.
Why do qPCR curves look abnormal in multiplex reactions?
Multiplex qPCR curves look abnormal when several targets compete for the same reaction components or when one assay performs better than the others. This can delay weak targets, reduce signal strength, or create uneven curve shapes.
Multiplex qPCR is useful because it can detect more than one target in the same well. But it also adds pressure to the reaction.
Each primer and probe set must work well with the others. A strong assay can dominate the reaction. A weak assay may amplify late or not at all.
In multiplex qPCR, a more efficient or more abundant target can suppress a weaker one because both reactions draw from the same pool of primers, probes, polymerase, nucleotides, and buffer components.
Each assay should prove itself alone before it is combined with other targets in the same well. Each assay should first show good efficiency, specificity, and clean curves alone. After that, the combined reaction can be adjusted.
A multiplex curve problem may be fixed by changing primer/probe concentrations, balancing targets, reducing template input, or adjusting cycling conditions. But if one assay is poorly designed, balancing alone may not solve it.
Why do qPCR curves look abnormal when the sample has inhibitors?
Inhibitors make qPCR curves late, weak, flat, or inconsistent. They interfere with polymerase activity, primer binding, reverse transcription, or fluorescence detection.
Different sample types carry different risks.
Blood may contain heme. Stool can contain bile salts and complex organic material. Soil and plant samples may contain humic acids or polyphenols. Tissue extraction can leave salts, ethanol, detergents, or phenol behind.
Too much template can also act like an inhibitor problem. Adding more sample does not always improve detection. Sometimes dilution gives a cleaner curve because it lowers inhibitor concentration more than it lowers target detection.
A simple dilution test can quickly show whether the sample is carrying substances that slow the reaction. If a diluted sample amplifies better than the undiluted sample, inhibition is likely.
An internal amplification control can also help. If the control target shifts late or fails in a sample, inhibition may be present.
A curve affected by inhibitors may still cross the threshold, but the Cq may not reflect the true starting amount. That is why inhibition checks are especially needed in clinical, environmental, forensic, and food-testing qPCR.
Why do qPCR curves look abnormal in RT-qPCR?
RT-qPCR curves can look abnormal because problems may happen before PCR even starts. RNA quality, reverse transcription efficiency, genomic DNA contamination, and sample handling all affect the final curve.
RNA is fragile. Heat, RNases, repeated freeze-thaw cycles, and poor storage can damage it.
When RNA is degraded, some targets may amplify poorly while others still work. Long amplicons are often affected more than short ones.
Reverse transcription adds another source of variation. If the enzyme, primer type, reaction temperature, or RNA input changes, cDNA yield may change too. That can cause late curves, scattered replicates, or false negatives.
Genomic DNA contamination can create early or unexpected curves, especially if primers do not span exon-exon junctions.
For RT-qPCR, controls become even more valuable. A no-RT control helps check genomic DNA contamination. A positive RNA control helps check the reverse transcription step. Reference genes help show whether sample input and quality are reasonably stable.
How does assay design cause abnormal qPCR curves?
Poor assay design can cause late amplification, nonspecific products, primer-dimers, weak signal, or no amplification. Even perfect pipetting cannot fully save a poorly designed primer or probe set.
Good qPCR assays usually have short amplicons, balanced primer melting temperatures, low primer complementarity, and strong target specificity.
Primer or probe mismatches can also hurt detection. This matters for pathogens, genetic variants, and any target with sequence variation. A mismatch near the 3′ end of a primer can reduce extension. A mismatch under a probe can weaken fluorescent signal or prevent probe binding.
Assay design issues may show up as:
- Low amplification efficiency
- Curves that rise late
- Different melt peaks
- Amplification in NTC wells
- Poor replicate agreement
- Weak fluorescence
- False negatives in some variants
The MIQE guidelines were created to improve qPCR reliability, transparency, and reproducibility across laboratories. They place strong weight on reporting assay details, sample quality, controls, and analysis choices because these details affect whether qPCR results can be trusted.
In practice, that means abnormal curves should not be treated as software problems alone. The assay itself may need review.
How do threshold and baseline settings create false abnormal curves?
Threshold and baseline settings can make a good curve look bad or a bad curve look acceptable. Software settings do not change the chemistry, but they can change the reported Cq.
If the threshold is too low, it may cross baseline noise. This can produce early or false Cq values.
If the threshold is too high, it may cross near the plateau. This can make Cq values late or inconsistent.
If the baseline range includes early amplification cycles, the software may subtract part of the real signal. If the baseline range is too short or too noisy, the threshold may be poorly placed.
Log view makes the exponential phase easier to judge, especially when the linear plot looks messy or compressed. In log view, the exponential phase is easier to see. A properly placed threshold should cut through the straight, rising part of the curve, not the flat baseline and not the plateau.
For publication-quality or regulated work, analysis settings should be consistent and recorded. Changing thresholds from plate to plate without a clear rule can create artificial differences.
How can you troubleshoot abnormal qPCR amplification curves?
The best way to troubleshoot abnormal qPCR curves is to read the pattern first, then check controls, replicate behavior, melt curves, sample quality, and analysis settings. Random changes waste time. Pattern-based troubleshooting gives faster answers.
Start with the controls.
If the positive control fails, look at reagents, cycling conditions, detector settings, and setup errors. If the NTC amplifies, check for contamination or primer-dimers. If only one sample behaves badly, check that sample for inhibitors, degradation, or loading error.
Next, compare replicates.
If one replicate is odd and the others are clean, a well-level issue may be likely. If all replicates are abnormal, the issue is probably shared across the assay or sample.
Then check the melt curve for SYBR Green assays. A single expected peak supports specificity. Extra peaks or low-temperature peaks suggest nonspecific products or primer-dimers.
After that, review baseline and threshold placement in log view.
If those settings are wrong, fix the analysis before repeating the experiment. If the curve shape itself is wrong, repeat the reaction with fresh reagents, clean setup, and proper controls.
A practical troubleshooting order looks like this:
- Check positive control, NTC, and extraction controls
- View curves in both linear and log formats
- Review threshold and baseline placement
- Compare technical replicates
- Check melt curves or product size
- Look for inhibition using dilution or internal control
- Review primer/probe design
- Repeat questionable samples with fresh setup
This order keeps you from blaming the assay when the real issue is software settings, or blaming the software when the chemistry failed.
When should an abnormal qPCR curve be rejected?
An abnormal qPCR curve should be rejected when it does not show true exponential amplification, appears in a contaminated control, has the wrong melt profile, differs sharply from valid replicates without explanation, or crosses the threshold because of noise.
A curve should not be accepted just because the software gives a Cq.
The Cq is only meaningful when the curve shape supports real amplification. A noisy line crossing the threshold is not the same as a valid amplification curve.
A result should be treated with caution or repeated when:
- NTC wells amplify with the same target profile
- Positive controls fail
- Melt curve shows the wrong product
- Replicates are widely scattered
- The curve crosses during baseline noise
- The sample shows signs of inhibition
- The plateau or exponential phase is missing
- The curve appears only after very late cycles without support from controls
Late Cq values need special care. Many labs set internal rules for late amplification, especially beyond 35–40 cycles, because late curves are more likely to involve low-copy randomness, primer-dimers, or contamination. The exact cutoff depends on assay validation, target type, and lab policy.
How can labs prevent abnormal qPCR curves?
Labs can prevent many abnormal qPCR curves through clean workflow, strong assay design, careful sample preparation, validated controls, and consistent analysis settings. qPCR is sensitive, so small habits make a big difference.
Physical separation helps reduce contamination. Pre-PCR setup, template handling, and post-PCR areas should be kept apart where possible.
Aerosol-resistant tips, fresh aliquots, clean benches, and regular decontamination reduce carryover risk.
Assay design should be checked before routine use. Primers and probes need specificity testing, efficiency testing, and control testing. Melt curves or probe specificity checks should be part of validation.
Sample quality also matters. Poor extraction can bring inhibitors into the reaction. Poor storage can degrade nucleic acids. Too much template can suppress amplification rather than improve it.
Consistent analysis settings matter too. Baseline and threshold choices should follow a repeatable rule, not guesswork. Reliable qPCR results depend on connected details such as sample quality, assay design, controls, efficiency, and reporting. When those details are handled carefully, abnormal curves are easier to catch before they lead to wrong conclusions.
What do abnormal qPCR curves usually mean in real lab work?
Abnormal qPCR curves usually mean the result needs review before interpretation. They do not always mean the experiment failed, but they do mean the curve should be checked against controls, replicate behavior, melt data, and assay expectations.
- A late curve may be a true low-copy target.
- A flat curve may be a true negative.
- A noisy curve may come from a bubble.
- An NTC curve may be primer-dimer rather than contamination.
- The curve alone gives a clue. The controls give the answer.
Experienced qPCR users usually look beyond the Cq table because the curve shape often reveals problems the number hides. They look at the curve shape first. Then they check whether the biology, chemistry, and controls all tell the same story.
A qPCR curve is not just a line on a screen. It is a record of how fluorescence becomes an amplification curve as the reaction behaves cycle by cycle. When that line looks abnormal, it is asking for a closer look before anyone trusts the number.