How Do Primers and Probes Affect Sequence Detection Results?
Primers and probes can make the difference between a clean sequence detection result and a confusing one. In real-time PCR or qPCR, primers decide what DNA or RNA target gets copied. Probes, when used, decide what signal gets reported. When they are designed well, the system can detect the right target with strong sensitivity, clear fluorescence, and reliable Ct or Cq values. When they are poorly designed, the result may show late amplification, false positives, weak signal, primer-dimers, or missed targets.
Primers control amplification, while probes control detection specificity and signal quality. A sequence detection system depends on both. Even a high-end instrument cannot fix a bad primer pair or a probe that binds poorly. That is why assay design, melting temperature, amplicon size, GC content, target region choice, and validation all matter before results can be trusted.
What role do primers play in sequence detection?
Primers are short DNA sequences that bind to the target region and give DNA polymerase a starting point for amplification. If the primers bind accurately, the correct target is copied. If they bind poorly, the reaction may fail or copy the wrong sequence.
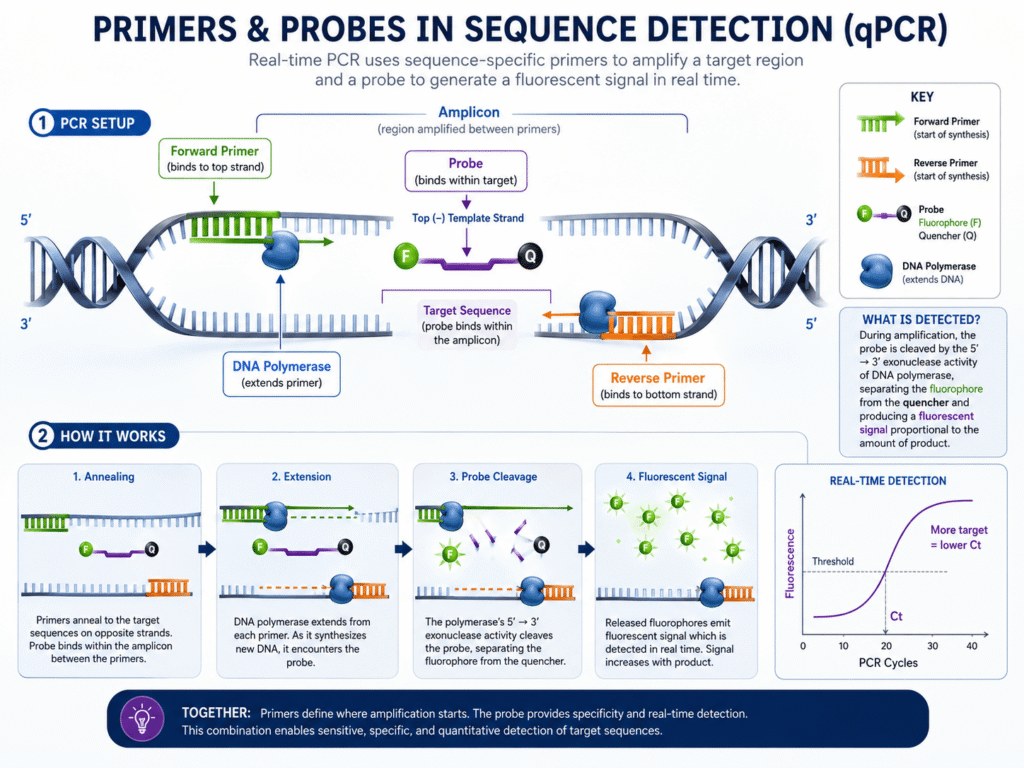
In qPCR and other sequence detection methods, primers sit at the center of the test. The forward primer binds to one strand, and the reverse primer binds to the opposite strand. The region between them becomes the amplicon.
A good qPCR assay should amplify the correct target with high specificity, avoid primer-dimers, and stay close to 100% amplification efficiency, according to qPCR assay design literature.
This matters because sequence detection systems do not “see” the original DNA in a direct way. They detect the signal produced as target copies increase. If the primers copy the wrong material, the system may still show a signal, but that signal may not represent the true target.
How do primers affect Ct or Cq values?
Primers affect Ct or Cq values by changing how fast the target amplifies. A strong primer pair gives earlier, cleaner amplification. A weak primer pair may delay amplification, raise Ct values, or create uneven results between repeated samples.
Ct, often called Cq in some systems, is the cycle number where fluorescence rises above the detection threshold. Lower Ct usually means more starting target. Higher Ct usually means less starting target or weaker amplification.
Poor primer design can make a sample look like it has less target than it really does. This happens when primers bind inefficiently, form secondary structures, or compete with primer-dimers.
Amplification efficiency and specificity depend heavily on both primer design and the target sequence, so even small design issues can change qPCR accuracy.
That is why two assays targeting the same gene can give different Ct values. The instrument may be the same. The sample may be the same. But a better primer pair can amplify faster and produce a clearer curve.
How does primer specificity affect sequence detection results?
Primer specificity affects whether the assay detects only the intended sequence or also amplifies similar, unwanted sequences. High specificity gives trustworthy results. Low specificity can create false positives or misleading quantification.
Specificity is especially important when targets belong to large gene families, viral variants, bacterial species, or closely related organisms. If primers bind to a similar sequence, the assay may report a positive result even when the intended target is absent.
Tools such as NCBI Primer-BLAST help check primer pairs against sequence databases, which reduces the chance of unwanted binding to the wrong template.
Specificity also matters in human gene expression work. Primers may accidentally amplify genomic DNA instead of cDNA if they are not designed with care. In RT-qPCR, many labs design primers across exon-exon junctions to reduce genomic DNA amplification, especially when RNA purity is not perfect.
What happens when primers form primer-dimers?
Primer-dimers form when primers bind to each other instead of the target sequence. They can create fluorescence, waste reaction components, and make the sequence detection result look stronger than it really is.
This problem is more visible in SYBR Green assays because SYBR Green binds to any double-stranded DNA. That includes the correct amplicon, nonspecific products, and primer-dimers.
Primer-dimers can form when primers bind to themselves or to each other, and labs often reduce this problem by adjusting primer concentration, raising annealing temperature, or redesigning the primer pair.
Primer-dimers often show up as extra peaks in melt curve analysis. They may also appear as late amplification in no-template controls. When that happens, the result should not be accepted without further review.
How does primer melting temperature affect results?
Primer melting temperature, or Tm, affects how strongly primers bind during the annealing step. If the Tm is too low, primers may bind nonspecifically. If it is too high or mismatched between primer pairs, amplification may become weak or uneven.
Many qPCR assays work best when primer Tm stays within a narrow range. A common target is about 58–60°C, with the forward and reverse primers kept very close to each other, often within about 1°C.
This small detail has a large effect on sequence detection. If one primer binds better than the other, amplification may become unbalanced. If both bind too loosely, nonspecific products may increase. If they bind too tightly, the reaction may lose efficiency.
Annealing temperature is also tied to Tm. Low annealing temperature can reduce specificity, so labs often raise it in small steps when nonspecific amplification appears.
How does primer concentration affect sequence detection?
Primer concentration affects amplification strength, background signal, and primer-dimer risk. Too little primer may cause weak or delayed amplification. Too much primer may increase nonspecific binding and primer-dimers.
Most assays need primer concentration testing before final use. A common range is tested experimentally because the best concentration depends on target sequence, master mix, probe chemistry, and reaction volume.
Probe qPCR kits commonly use primer concentrations within a tested range, often around 0.05 to 1 µM, while probe concentration is adjusted based on the assay’s signal and background.
The best concentration is not always the highest one. A lower primer concentration may give cleaner results if dimers or nonspecific products are appearing. The goal is a strong signal from the right target, not just the earliest possible Ct.
What role do probes play in sequence detection?
Probes add another layer of target recognition. In probe-based qPCR, the probe binds inside the amplicon, between the forward and reverse primers. Fluorescence is released only when the correct target region is amplified and the probe is cleaved or activated.
This is why probe-based assays, such as TaqMan assays, are often chosen for clinical testing, pathogen detection, genotyping, and multiplex assays. The primers must bind correctly, and the probe must also bind correctly.
Probe chemistries add sequence-specific detection to real-time PCR, while dye-based methods can produce signal from any double-stranded DNA product.
That added recognition step can reduce false signal from primer-dimers. A primer-dimer may still form, but it usually will not produce probe fluorescence unless the probe also binds to it, which is unlikely when the assay is well designed.
How do probes affect specificity?
Probes raise specificity because they must bind to a target sequence inside the amplified region. This means the assay depends on three sequence-binding events: forward primer binding, reverse primer binding, and probe binding.
This is a major reason hydrolysis probe assays are often preferred when a test must distinguish between closely related targets. A single mismatch in the probe region can reduce signal, especially if the mismatch sits near the middle of the probe.
Dye-based detection can report fluorescence from nonspecific double-stranded DNA, while probe-based detection gives stronger specificity because the signal depends on target-specific probe binding.
This does not mean probes make an assay perfect. A badly placed probe can still bind weakly, miss variants, or fail in multiplex reactions. But in general, probes help the sequence detection system report the intended target more selectively.
How does probe melting temperature affect results?
Probe melting temperature affects how reliably the probe binds during amplification. A probe usually needs a higher Tm than the primers so it stays bound while extension occurs.
For Applied Biosystems TaqMan-style assays, probe Tm is commonly designed about 10°C higher than primer Tm so the probe binds firmly during amplification.
This higher Tm helps the probe bind firmly to the target before polymerase reaches it. If the probe Tm is too low, fluorescence may be weak because the probe does not stay attached long enough. If the Tm is too high or the probe has poor sequence balance, background signal or poor quenching can become a problem.
Probe chemistry also matters. Minor groove binder probes can have higher Tm with shorter sequences, which can help when the target region is short or contains mutations that must be detected with high precision.
How do probe mismatches affect sequence detection?
Probe mismatches can reduce fluorescence, delay Ct values, or cause false negatives. This happens when the target sequence has a mutation, variant, or polymorphism where the probe is supposed to bind.
This issue became widely discussed during infectious disease testing, where viral genomes can change over time. A primer or probe designed for an older sequence may not bind as well to a newer variant if mutations appear in the binding region.
A mismatch does not always destroy an assay. Its effect depends on location, number of mismatches, probe length, chemistry, and reaction conditions. But even one poorly placed mismatch can reduce signal enough to change interpretation in low-copy samples.
For diagnostic assays, this is why labs often check primer and probe sequences against current databases and monitor whether target organisms have changed in the binding regions.
How does amplicon size affect sequence detection?
Amplicon size affects amplification speed and efficiency. Shorter qPCR amplicons usually amplify more efficiently, especially when the sample is degraded or present at low levels.
A practical qPCR design range is often a 70–150 bp amplicon with primer GC content around 40–60%.
Short amplicons are helpful because qPCR measures amplification in real time, not just at the end. A smaller product can be copied faster and more consistently. This is valuable in clinical samples, formalin-fixed tissue, environmental samples, and old RNA, where nucleic acids may be fragmented.
Long amplicons may still work, but they often reduce sensitivity. They can also make results less consistent when sample quality varies.
How do primers and probes affect sensitivity?
Primers and probes affect sensitivity by controlling how few target copies the assay can detect. Good binding, short amplicon length, clean chemistry, and low background help the system detect low-copy targets.
Sensitivity matters most near the limit of detection. A sample with many target copies may still amplify even with a mediocre assay. A sample with very few copies may be missed if primer binding is weak or probe signal is poor.
qPCR can detect and measure very small amounts of nucleic acid, but the result only carries weight when assay design, reporting, and validation are handled carefully.
In real lab work, sensitivity is not proven by design alone. It must be tested with dilution series, positive controls, negative controls, and replicate reactions. A primer-probe set that looks good on paper may still perform poorly with real samples.
How do primers and probes affect false positives?
Primers and probes can cause false positives when they bind unintended targets, produce primer-dimers, or generate background fluorescence. This can make a negative sample appear positive.
False positives are more common when primer specificity is poor, annealing temperature is too low, or contamination is present. In SYBR Green assays, any double-stranded DNA can create signal, so melt curve analysis becomes very useful.
Probe assays lower this risk because the probe must also bind to the amplified region. Still, probe assays can produce false positives if contamination from a true positive sample enters the reaction.
No-template controls are used to catch this problem. If the control amplifies, the run needs review before results are reported.
How do primers and probes affect false negatives?
False negatives can happen when primers or probes do not bind the target well enough. This may be caused by mutations, poor design, low template amount, inhibitors, degraded sample, or incorrect reaction conditions.
A false negative is more dangerous in diagnostic testing because it may tell a patient or clinician that a target is absent when it is present.
Primer or probe mismatches are one cause. Another cause is poor target choice. If the assay targets a low-copy or unstable region, detection may be weaker than an assay targeting a more conserved region.
For RNA viruses, bacterial subtypes, or genetically diverse organisms, target region selection is one of the most important design choices. Conserved regions reduce the chance that natural sequence variation will block binding.
How do primers and probes affect amplification efficiency?
Amplification efficiency shows how well the target doubles during each PCR cycle. Good primer-probe sets produce efficiency close to ideal, while poor sets create slow, uneven, or unreliable amplification.
In qPCR, efficiency is often tested using a standard curve. A clean assay usually gives a straight standard curve across serial dilutions. If the curve bends, spreads, or gives strange slopes, the assay may not be reliable across different target amounts.
qPCR assay design can perform differently in real samples than it does in theory, which is why reporting, testing, and validation matter so much.
Efficiency problems can come from primer secondary structure, target folding, GC-rich regions, poor amplicon length, bad probe placement, or reaction chemistry. Primer-probe design should never become a copy-and-paste step.
What is the difference between primers and probes in result quality?
Primers affect whether the target is amplified. Probes affect whether the amplified target is detected with added specificity. Both shape result quality, but they do different jobs.
A primer problem often appears as no amplification, late Ct, multiple products, or primer-dimers. A probe problem often appears as weak fluorescence, poor signal separation, abnormal curves, or failure to detect known positives.
In SYBR Green assays, there are primers but no target-specific probe. The signal comes from dye binding to double-stranded DNA. In TaqMan-style assays, the probe creates a target-specific fluorescence signal during amplification.
This makes probe assays more expensive and more complex to design, but often better for multiplexing and high-specificity testing.
How do primers and probes affect multiplex sequence detection?
In multiplex assays, several primer-probe sets run in the same tube. Each set must amplify its own target without interfering with the others. Poor design can cause competition, weak signals, or mixed fluorescence patterns.
Multiplexing is useful because one reaction can test several targets at once. For example, a respiratory panel may test for multiple viruses, or a gene expression assay may measure a target gene and a reference gene together.
The challenge is balance. One primer pair may amplify faster and consume reagents before a weaker target can amplify. Probes also need different fluorescent dyes with limited spectral overlap.
This is why multiplex assays need more validation than single-target assays. Each target must work alone, then work together in the combined reaction.
How do primers and probes affect infectious disease detection?
Primers and probes affect infectious disease detection by deciding whether the test can find the pathogen accurately in real samples. Good design can detect low pathogen loads. Poor design can miss infections or cross-react with related organisms.
For viruses, bacteria, fungi, and parasites, the target region must be specific enough to avoid other organisms but conserved enough to remain stable across strains.
A test for a fast-changing virus may need regular sequence checks. A test for a bacterial species may need checks against closely related species. A test for a parasite may need special care if the target region appears in more than one organism.
For public health and diagnostic labs, primer-probe design is part of assay quality, not just a technical setup step.
How can labs tell if primer and probe design is working?
Labs can tell by checking amplification curves, Ct consistency, standard curve efficiency, melt curves when using dyes, no-template controls, positive controls, and results across dilution series.
A working assay usually shows:
- Clear amplification in positive samples
- No amplification in no-template controls
- Consistent Ct values between replicates
- Good standard curve behavior
- No extra melt curve peaks in dye-based assays
- Strong signal separation from background
- Expected results with known positive and negative samples
These checks help separate true biological results from assay noise. A clean-looking curve is helpful, but it is not enough on its own. The assay must show that it detects the right target at the right level.
What are the most common primer and probe design mistakes?
The most common mistakes include choosing a poor target region, ignoring sequence variation, using mismatched primer Tm values, designing amplicons that are too long, allowing primer-dimers, and skipping validation.
Some mistakes are easy to miss. A primer may pass a basic design tool but still bind a related organism. A probe may look fine but sit over a mutation hotspot. A primer pair may work in a pure control but fail in real samples with inhibitors.
Good design starts with sequence selection. Then it moves through software checks, specificity screening, concentration testing, efficiency testing, and validation with real sample types.
What makes a strong primer-probe set?
A strong primer-probe set binds only the intended target, amplifies efficiently, avoids primer-dimers, gives clean curves, and performs well across low and high target amounts.
Good primer-probe sets usually have balanced Tm values, reasonable GC content, short amplicons, no strong secondary structures, and strong specificity against related sequences. Probe placement should avoid known mutation sites whenever possible.
The final proof is performance. A primer-probe set is not strong because software says so. It is strong when test data show that it gives accurate, repeatable, and specific results.
Why primers and probes are the quiet drivers of reliable detection
A sequence detection system may look like the star of the process, but primers and probes often decide the result before the run begins. They guide the reaction, shape the signal, and influence whether the answer is clear or questionable.
When primers are specific and efficient, amplification reflects the real target. When probes are well placed and well matched, fluorescence reflects the right sequence. Together, they turn raw nucleic acid into a result a researcher, clinician, or lab team can trust.
Better primer and probe design does not just improve a graph on a screen. It protects the meaning behind the result. That is what makes assay design one of the most valuable steps in sequence detection.